|
|
. |
Project 1. Preparation of 1- and 2-Adamantanol and Assignment of Proton NMR Spectra Techniques: Multistep organic synthesis, recrystallization, extraction, column chromatography, proton NMR spectroscopy, IR spectroscopy, lanthanide shift reagents, use of chemical literature, graphics Adamantane is a particularly interesting system for NMR analysis. Although the structural formula does not reveal its high symmetry to the casual observer, the molecule has tetrahedral symmetry. Application of these symmetry operations reveals that adamantane has only two types of carbon atoms and, consequently, only two types of hydrogens. The rigidity of the structure also has several consequences. The vicinal proton coupling usually observed in other molecules is very weak in adamantane derivatives because of the Karplus dihedral angle-coupling constant relationship. Secondly, the internal rotational motions that sometimes affect shifts and couplings are in this case absent. In order to make use of a lanthanide shift reagent for spectral simplification, a suitable functional group must be present on the adamantane skeleton. The OH group is particularly appropriate in this case because of the relatively easy preparation of the alcohols. 1-Adamantanol is prepared by bromination of adamantane, followed by nucleophilic substitution with hydroxide ion, while 2-adamantanol is secured by oxidation of adamantane with sulfuric acid, followed by reduction with sodium borohydride. Preparation of 1-Bromoadamantane. This reaction must be conducted in a hood using great care (gloves and goggles) in the handling of bromine and carbon tetrachloride. Slowly add 25 mL of bromine to 10 g of adamantane in a 100-mL, one-neck, round-bottom flask containing a magnetic stir bar and condenser. Gently heat the mixture for 1 h, and then gradually increase the heat and maintain a mild reflux for another hour. Cool the mixture, and dissolve it in carbon tetrachloride. Wash the resulting solution with 100-mL portions of a saturated aqueous solution of NaHSO3 until the solution is decolorized. Wash the solution several times with water, and then dry the carbon tetrachloride layer over magnesium sulfate. Remove the carbon tetrachloride layer under vacuum, and recrystallize the 1-bromoadamantane from a minimum amount of methanol. Obtain the melting point of the product and compute the percentage yield. Preparation of 1-Adamantanol. Mix 10 g of 1-bromoadamantane with 15 mL of 2 M HCl(aq) and 25 mL of dimethylformamide in a 100-mL, one-neck, round-bottom flask equipped with stir bar and condenser. Heat the mixture gently and stir magnetically to dissolve the materials, and then heat at 110 oC for two hours. Any 1-bromoadamantane that sublimes to the upper surface of the flask must be retained by gentle swirling of the solution. Cool the flask, add 20 mL of water, and allow the mixture to remain overnight. Suction filter the crystals and wash them with water. Determine the yield and melting point of the dry crystals, and obtain the infrared spectrum of the product (do not oven dry). If the melting point of the product is not sharp, purify the compound by column chromatography on alumina. Preparation of 2-Adamantanone. Concentrated sulfuric acid (140 mL) is added to 7.0 g of finely divided adamantane in a 250-mL, single-neck, round-bottom flask equipped with a magnetic stir bar. Stopper the flask loosely (SO2(g) is released during the reaction). Stir the contents, and heat the mixture to 77 oC in an oil bath for 6 h. Agitate the flask every half hour to disperse sublimed adamantane. The reaction mixture will turn black after the adamantane is completely dissolved. Cool the mixture to room temperature, and then pour it over 400 g of ice. Place the mixture under a hood, and add ether. Separate the ether layer using a separatory funnel. Extract the aqueous layer again. Also extract the black residue in the flask twice with ether. Combine and evaporate all ether extracts. Redissolve the residue in a minimal amount of ether. Wash this solution with a saturated sodium chloride solution, and then dry the ether layer over sodium sulfate. Filter the dried ether layer, and collect the product after evaportation of the ether. Check a sample of the solid for unreacted adamantane by dissolving a small portion in methylene chloride and obtaining a proton NMR spectrum. If unreacted adamantane is present, remove it by column chromatography on alumina. Dissolve the sample in a minimal amount of methylene chloride, and elute it with toluene. Adamantane will be eluted first, and it sublimes easily, particularly during evaporation of the toluene. Obtain the yield and melting point of the crude and pure product. Preparation of 2-Adamantanol. Place a solution of 4.0 g of 2-adamantanone in 50 mL of isopropyl alcohol in an Erlenmeyer flask. While stirring this solution magnetically, slowly add 1.6 g of sodium borohydride. Continue the stirring for 30 minutes after addition. Add a 50-mL portion of ether, followed by a slow addition of a 10% HCl(aq) solution. After stirring the solution for several minutes, add the same amount of a saturated NaHCO3 solution. Separate the ether layer, and dry it over magnesium sulfate. Remove the solvent under vacuum, and check the remaining solid for a carbonyl absorption in the infrared. If the absorption is present, subject the solid to another reaction with sodium borohydride under reflux, Obtain the melting point and yield of 2-adamantanol. NMR Analysis. After becoming thoroughly familiar with all aspects of the operation of the Varian EM-360A proton NMR spectrometer, obtain the spectrum of a solution of 60.0 mg of l-adamantanol in 1.0 mL of CDCl3. A trace amount of TMS may be added as an internal reference, but this can cause complications. The small amount of residual CHCl3 present in the deuterated solvent is normally sufficient to serve as a standard that can be used to estimate TMS-based chemical shifts. Add 40.0 mg of solid Eu(fod)3 shift reagent to the adamantanol solution previously prepared. Compute the shift reagent/ adamantanol molar ratio, and obtain the proton NMR spectrum at sweep widths of both 10 and 20 ppm. Add an additional 40.0 mg of solid shift reagent to the sample tube, compute the new shift reagent/adamantanol molar ratio, and record two more spectra. Repeat this procedure at least two additional times, each time using the previous solution with an additional 40.0-mg increment of shift reagent. Plot all sets of proton chemical shifts for all protons in the sample versus the corresponding molar ratio (the ratio for the original unshifted sample will be 0.0). Assign all protons, and repeat the entire procedure for a 60-mg sample of 2-adamantanol. Your report should contain discussions
of: 1) the probable nature of the reaction mechanisms of the formation
of the alcohols supported by literature citations; 2) the reasoning involved
in the choice of alumina as the stationary phase in the chromatographic
purifications of the compounds; and 3) the assignments of the shifted peaks
in the proton NMR spectra of the alcohols, supported by distances from
europium to each different type of hydrogen atom, measured from scale models
of the compounds. Use the following distances (center of atom to center
of atom) and angle in constructing your models: r(Eu-O) = 251 pm, r(C-O)
= 143 pm, r(C-C) = 154 pm, r(C-H) = 109 pm, angle (Eu-O-C) = 115o.
Assume that the Eu-O bond is freely rotating, and use the shortest distance
from Eu to each hydrogen.
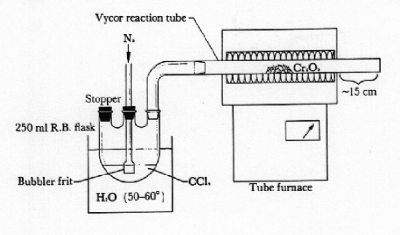
1-Adamantanol 2-Adamantanol Project 2. Preparation of Anhydrous Chromium(III) Chloride Techniques: High temperature synthesis, tube furnance, inert atmosphere High temperature synthesis is an excellent method of preparing many anhydrous transition metal chlorides. Some of these materials possess such great affinity for water that the anhydrous compounds canot be sold commercially. Anhydrous chromium(III) chloride may be obtained by treating fresh chromic oxide with carbon tetrachloride vapor at 650 oC. Cr2O3(s) + 3 CCl4(l) = 2 CrCl3(s) + 3 COCl2(g) This reaction is conducted in a tube furance under a nitrogen atmosphere. Although anhydrous chromic oxide may be purchased from several commercial sources, better yields on the high temperature conversion are obtained when using freshly prepared oxide. The spectacular thermal decomposition of (NH4)2Cr2O7 is a quick way to secure dry chromic oxide. (NH4)2Cr2O7(s) = Cr2O3(s) + N2(g) + 4 H2O(l) Preparation of Chromium(III) Oxide. Place 4 g of ammonium dichromate in the center of a large porcelain dish in a hood. Touch a weak Bunsen flame the material until the reaction proceeds by itself. Hot solids and sparks will rapidly be thrown into the air. After the reaction has stopped, extract the green insoluble product with hot distilled water until the washings are colorless. Dry the chromic oxide at 100 oC, and determine the yield of the reaction. Preparation of Chromium(III) Chloride. In a hood, set up the tube furnace and related apparatus as shown in the figure below. The reaction tube must be able to withstand a temperature of at least 800 oC. Regular Pyrex™ glass is unsatisfactory; Vycor™ brand glass tubing should be used. Place 1.5 g of chromium(III) oxide in the center of the reaction tube. Add sufficient carbon tetrachloride to cover the bubbler frit in the round-bottom flask. Heat the water bath to 55 oC with a hot plate and set the tube furnace to 800 oC. Begin bubbling a slow stream of nitrogen gas through the carbon tetrachloride so that gentle purging is maintained in the round-bottom flask (too rapid a nitrogen flow rate will blow the chromic oxide powder out of the reaction tube). Turn on the tube furnace, and allow it to slowly climb to the 800 oC reaction temperature. This reaction will require at least 1 1/2 to 2 h at 800 oC for completion. When no green chromic oxide remains, remove the water bath, raise the bubbler above the carbon tetrachloride surface, and turn off and open the tube furnace. Allow the product to cool to room temperature under a slightly increased nitrogen flow rate. Weigh the product and determine the percentage yield. Store the product in a screw-cap bottle. Your report should contain a discussion of: 1) preparations (with and/or without high temperature conditions) for at least three other anhydrous transition metal halides (including balanced equations); 2) why it is desirable to maintain a nitrogen atmosphere in your reaction; and 3) several methods of characterizing chromium(III) chloride.
Project 3. Preparation of Dicarbonyl(h5-methylcyclopentadienyl)triphenylphosphinemanganese Techniques: Transition metal organometallic photochemistry, IR spectroscopy, recrystallization Carbonyl complexes of transition metals serve as starting materials for many chemical reactions in inorganic and organometallic chemistry. The substitution of a coordinated carbonyl ligand can often be induced by light of the proper frequency, according to the following reaction. To increase yields, the M(CO)n + L zz/M(CO)n-1L + CO(g) photochemical reaction is often carried out in tetrahydrofuran (THF) or diethyl ether. The transition state is believed to be stabilized in these weakly coordinating solvents to afford an observable M(CO)n-1(solvent) intermediate. Addition of a ligand, L, then yields the product by replacement of the coordinated solvent molecule, according to the following pathway. M(CO)nzz/M(CO)n-1(solvent) zz/ M(CO)n-1L The compound tricarbonylmethylcyclopentadienylmanganese, which has been widely used as an octane-booster in unleaded gasoline, possesses a rich carbonyl photosubstitution chemistry, often to yield CH3C5H4Mn(CO)2L, where L = olefins, amines, phosphines, sulfur dioxide, etc., by the two routes shown in the scheme. We are interested in conducting the reaction in an inert atmosphere in n-hexane when L is triphenylphosphine. CH3C5H4Mn(CO)3zzzzzzz/
CH3C5H4Mn(CO)2L
CH3C5H4Mn(CO)2(THF) + CO(g) Preparation of Dicarbonyl(h5-methylcyclopentadienyl)triphenylphosphinemanganese. Distill 25 mL of (h5-methylcyclopentadienyl)tricarbonylmanganese under vacuum using a water aspirator, 6-inch Vigreux column, and an Ace Mini-Lab® distillation head. Collect a fraction which boils at 130-132 oC at 32 torr. Charge a 500-mL Erlenmeyer flask with 200 mL of n-hexane, 2 mL of freshly distilled CH3C5H4Mn(CO)3, and 4.0 g of triphenylphosphine. Deaerate the vessel with argon or nitrogen, and place the flask in direct sunlight. A slow stream of inert gas should pass through the flask for four days. During this time, swirl the contents of the flask several times each day. Periodically, monitor the IR spectrum of the solution in the CO stretching region (2100-1800 cm-1), checking for diminished intensity of CO peaks in the starting material and enhancement of product CO peaks. Filter the hexane to obtain a small quantity of red crystals. Recrystallize the product by dissolving it in hot acetone, followed by cooling to 0o C with addition of 5-10 drops of distilled water. The product may also be recrystallized from ethanol. The melting point of the product is reported to be 119-120 oC. Your report should contain a discussion
of: 1) the assignments of the IR bands in the product and starting materials;
2) identification of any other possible substitution products which may
form in the reaction; and 3) physical properties of the product. Also,
discuss commercial uses of the starting manganese compound.
Project 4. Preparation, Spectroscopy and Reactivity of Bis[N,N-bis(trimethylsilyl)amino]tin(II) Techniques: Dry box, glove bag, vacuum line, syringe and needle methods, organolithium reagents, vacuum distillation, UV, IR, and NMR spectroscopy One of the curious observations concerning the chemistry of many of the main group elements is that stable alkyl or dialkylamine derivatives exist only for those elements which have closed-shell electron configurations. This is almost invariably true even when the element in question displays more than one valence. For example, the stable derivatives of tin or lead are species such as Sn(CH3)4, Sn[N(C6H5)2]4, or Pb(C2H5)4, but not the corresponding divalent compounds Sn(CH3)2, Sn[N(C6H5)2]2, and Pb(C2H5)2. Although many divalent halogen derivatives of tin and lead are well known, examples of stable, monomeric, divalent tin and lead compounds containing direct carbon or nitrogen attachments are extremely rare. The first stable, monomeric, divalent tin-nitrogen derivatives resulted in an additional surprise: the compounds are intensely colored. Color is extremely rare for monomeric compounds of the main group elements. The striking red color of the title compound is even more unusual when recognizing that the N[Si(CH3)3]2 ligand, when incorporated in compounds of many other elements, does not generally impart color. Preparation of Bis[N,N-bis(trimethylsilyl)amino]tin(II). Set up a three-neck, 250 mL, round-bottom flask fitted with a condenser, pressure-equalizing dropping funnel, magnetic stir bar, and inert gas inlet. All equipment must be dried in a 110 oC oven for several hours. A solution of hexamethyldisilazane (7.4 g) in anhydrous tetrahydrofuran (30 mL) is placed in the dropping funnel. With an oven-dried syringe, carefully transfer 20 mL of a 2.4 M n-hexane solution of n-butyllithium to the three-neck flask by injecting it through a rubber septum placed in the center neck. Cool the flask to 0 oC using an ice/water bath, and carefully increase the inert gas flow rate to prevent oil from the bubbler from pulling back into the reaction vessel. After 15 minutes of magnetic stirring, begin a slow dropwise addition of the amine solution to the organolithium reagent. After addition is complete, allow the yellow solution to warm to room temperature. Prepare a solution of anhydrous tin(II) chloride (4.7 g) in 40 mL of dry tetrahydrofuran, and add this solution dropwise to the lithiated amine. Stir the reaction mixture at room temperature during addition, and for 2 h after the addition is complete. Filter the dark reaction mixture through Celite® in a glove bag fitted for simultaneous inert gas purging and suction filtration. The Celite® should also be dried at 110 oC prior to use. Remove solvent from the resulting solution at reduced pressure, so that a minimum amount of heat is applied to the sample. Distill the remaining crude liquid on a 6-inch column packed with glass beads. A pressure of 4-10 torr is adequate to prevent substantial decomposition of the product. Collect at least three fractions. The sample is highly sensitive to trace amounts of oxygen and moisture. Compute the yield on this reaction. Record your observations when oxygen, water, and methanol are separately reacted with small portions of the purified tin amine. Freeze another small portion of the sample and record any unusual observations. Obtain the UV (neat), IR (liquid film), and proton NMR (in benzene-d6) of the tin amine, and also record the IR and proton NMR spectra of hexamethyldisilazane for comparison purposes. Your report should contain a discussion
of: 1) reactivity and spectroscopic data for the tin amine, including an
assignment of the tin(II)-nitrogen stretching frequency in the infrared
spectrum; and 2) the physical properties of the substance.
Project 5. Preparation, Analysis, Spectroscopy, and Lewis Acidity of Tin(II) and Tin(IV) Iodide Techniques: Inert atmosphere, direct synthesis, gravimetric tin analysis, and NMR spectroscopy Obtain a copy of the article describing this work (R.W. Schaeffer, B. Chan, M. Molinaro, S. Morissey, C.H. Yoder, C.S. Yoder, and S. Shenk, J. Chem. Educ., 74, 575 (1997)) and carry out the syntheses of tin(II) iodide and tin(IV) iodide. Preparation of Tin(II) Iodide From the Elements. Place a 0.5-g sample of tin and a 1.2-g sample of iodine into a three-neck, 100-mL flask fitted with a cold-water condenser and a mineral oil bubbler. Thoroughly flush the flask with nitrogen for fifteen minutes. Next, add a 10-mL aliquot of 2 M HCl to the vessel. Heat to gentle reflux for an hour until all iodine has reacted. Gravity filter the solution while hot into another three-neck flask to remove unreacted tin. Keep the flask hot and repurge with nitrogen for 10-15 minutes to remove oxygen introduced while filtering. Collect the resulting crystals, formed by cooling the mixture in an ice bath under nitrogen, by vacuum filtration. Wash the product with 10 mL of cold water and allow it to dry in a vacuum desiccator. Store the product in a vacuum desiccator backfilled with nitrogen gas to prevent reaction with atmospheric oxygen. Obtain a melting point and compare with a literature value. Preparation of Tin(IV) Iodide From the Elements. Place 1.0 g of tin and 2.5 g of iodine into a 50-mL Erlenmeyer flask along with 15 mL of toluene. Cover the flask with a watch glass and heat gently on a hot-plate until the toluene begins to boil. Reduce the heat immediately (the reaction is exothermic and requires little heat once started). Gently heat the flask with occasional swirling for 20-30 minutes until the violet color of iodine disappears. Filter the hot solution into a 50-mL beaker and cool it in an ice bath. Collect the bright orange crystals by vacuum filtration. Wash the crystals with 5 mL of cold toluene, and allow them to dry in a vacuum desiccator. Recrystallize the product from a minimum amount of cold toluene and obtain the melting point after drying. Preparation of Tin(IV) Iodide From Tin(II) Iodide. Place samples of SnI2 (0.20 g and I2 (0.14 g) in a 100-mL, three-neck flask fitted with a cold-water condenser and mineral oil bubbler. Flush the system with nitrogen for fifteen minutes. Add a 20-mL aliquot of toluene to the system and heat the reaction mixture to reflux until all evidence of violet iodine disappears. Filter the hot solution into a 150-mL Erlenmeyer flask and cool it in an ice bath. Collect the resulting large orange-red crystals and recrystallize them from toluene. Determine the melting point and compare with your earlier result for the tetraiodide. Preparation of Sodium Hexaiodostannate(IV), Na2[SnI6]. Dissolve 1.00 g of SnI4 and 0.50 g of NaI with gentle heating in 20 mL of acetone in a 100-mL beaker. A dark, almost black, solution forms. Allow this solution to cool and place it in a desiccator overnight to permit the acetone to slowly evaporate. Iridescent dark-blue crystals result which decompose promptly. Gravimetric Analysis of Tin Compounds. Obtain three porcelain crucibles and heat them to constant weight using a Bunsen burner. Place approximately 0.5 g (weighed to the nearest 0.1 mg) of SnI4 into each crucible followed by 1 mL (20 drops) of 3 M HNO3. Cover each crucible and leave undisturbed for at least 24 h. Add an additional 10 drops of 3 M HNO3 and allow the crucibles to remain for several more hours. Gently heat the crucibles on a hot-plate for several hours followed by heating with a low Bunsen flame to remove volatiles (avoid splattering). Finally, heat the crucibles strongly over a Bunsen flame for 30 minutes, allow them to cool for several minutes, and place them in a desiccator until they reach room temperature. Weigh the crucibles and contents and calculate the composition of the original SnI4 assuming complete conversion to SnO2. Repeat this procedure for SnI2 using longer, more gentle digestion periods with stirring. NMR Parameters. Obtain tin-119 NMR spectra on saturated solutions in 10-mm O.D. tubes containing a coaxial inner tube with a 0.1 M solution of tetra(n-propyl)tin in acetone-d6. Your report should contain a discussion
of: 1) reactivity, analytical, and spectroscopic data for the tin-iodine
compounds, including assignments of the resonances in the tin-119 NMR spectra
and comparisons with literature values; and 2) the physical properties
of the substances.
References Project Reference 1 R.C. Fort, Jr., and P. Von R. Schleyer, Chem. Rev., 64, 277 (1964). F.W. Van Deursen and P.K. Korver, Tetrahedron Lett., 3923 (1967). A.F. Cockerill and D.M. Rackham, Tetrahedron Lett., 5149 (1970). G.H. Wahl, Jr., and M.R. Peterson, Jr., Chem. Commun., 1167(1970). J.R. Campbell, Aldrichim. Acta, 4, 55 (1971). R.S. Monson, "Advanced Organic Synthesis," Academic Press, NY, 1971, pp. 152-153. M.R. Peterson, Jr., and G.H. Wahl, Jr., J. Chem. Educ., 49, 790 (1972). R. Von Ammon and R.D. Fischer, Angew. Chem., Int. Ed. Engl., 11, 675 B.C. Mayo, Chem. Soc. Rev., 2, 49 (1973). A.F. Cockerill, G.L.O. Davies, R.C. Harden, and D.M. Rackham, Chem. Rev., 73, 553 (1973). R.E. Sievers, Ed., "Nuclear Magnetic Resonance Shift Reagents," Academic Press, NY, 1973. R.C. Fort, Jr., "Adamantane: The Chemistry of Diamond Molecules," Marcel Dekker, NY, 1976. K.A. Kime and R.E. Sievers, Aldrichim. Acta, 10, 54 (1977). C.D. Schaeffer, Jr., and C.H. Yoder, J. Chem. Educ., 62, 537 (1985). T.C. Morrill, Ed., "Lanthanide Shift Reagents in Stereochemical Analysis," VCH Publishers, Deerfield Beach, FL, 1986 (Methods in Stereochemical Analysis, Vol. 5). T.J. Wenzel, Ed., "NMR Shift Reagents,"
CRC Press, Boca Raton, FL, 1987.
2 R.J. Angelici, "Synthesis and Technique in Inorganic Chemistry," 2nd ed., W.B. Saunders Company, Philadelphia, PA, 1977, pp. 33-37 (currently available from University Science Books, Mill Valley, CA). A. Vavoulis, T.E. Austin, and S.Y. Tyree, Inorg. Syn., 6, 129 (1960). C. Stevenson and R. Rudman, J. Chem.
Educ., 71, 704 (1994).
3 C. Barbeau, Can. J. Chem., 45, 161 (1967). A.R. Manning, J. Chem. Soc. A, 106 (1971). D.C. Calabro and D.L. Lichtenberger, J.
Chem. Educ., 59, 686 (1982).
4 C.D. Schaeffer, Jr., and J.J. Zuckerman, J. Am. Chem. Soc., 96, 7160 (1974). D.H. Harris and M.F. Lappert, J. Chem. Soc., Chem. Commun., 895 (1974). P.J. Davidson, D.H. Harris, and M.F. Lappert, J. Chem. Soc., Dalton Trans., 2268, 2275 (1976). M.F. Lappert and P.P. Power, in "Organotin Compounds: New Chemistry and Applications," J.J. Zuckerman, Ed., Advances in Chemistry Series No. 157, American Chemical Society, Washington, DC, 1976, Ch. 5. M.J.S. Gynane, D.H. Harris, M.F. Lappert, P.P. Power, P. Rivière, and M. Rivière-Baudet, J. Chem. Soc., Dalton Trans., 2004 (1977). M.F. Lappert, P.P. Power, M.J. Slade, L. Hedberg, K. Hedberg, and V. Schomaker, J. Chem. Soc., Chem. Commun., 369 (1979). M.F. Lappert, P.P. Power, A.R. Sanger, and R.C. Srivastava, "Metal and Metalloid Amides," Wiley-Halsted, NY, 1980, Ch. 5. T. Fjeldberg, H. Hope, M.F. Lappert, P.P. Power, and A.J. Thorne, J.Chem. Soc., Chem. Commun., 639 (1983). C.D. Schaeffer, Jr., L.K. Myers, S.M. Coley,
J.C. Otter, and C.H. Yoder, J. Chem. Educ., 67, 347 (1990).
5 T. Moeller and D.C. Edwards, Inorg. Synth., 4, 119 (1953). G.G. Hickling, J. Chem. Educ., 67, 702 (1990). R.W. Schaeffer, B. Chan, M. Molinaro, S.
Morissey, C.H. Yoder, C.S. Yoder, and S. Shenk, J. Chem. Educ.,
74,
575 (1997).
|
|
|
Copyright © 1998 Elizabethtown College All Rights Reserved Maintained by Charles D. Schaeffer |